先行バイオ医薬品との同等性
腎性貧血を有するHD患者を対象とした、先行バイオ医薬品との同等性を検証した臨床成績を紹介します。
透析患者への有効性・安全性
腎性貧血を有するHD患者を対象とした、本剤の有効性と安全性を紹介します。
保存期慢性腎臓病(ND)患者への有効性・安全性
ESA治療中のNDの腎性貧血患者を対象とした、本剤の有効性と安全性を紹介します。
01 先行バイオ医薬品との同等性
本試験は治療期1期及び2期から成り、構成及び検討内容は以下の通りであった。
- 治療期1期[同等性検証試験]:ヘモグロビン(Hb)濃度変化量により、本剤と先行バイオ医薬品との同等性を検証した。また、安全性も検討した。
- 治療期1期及び治療期2期[長期投与試験]:本剤の安全性及び有効性を検討した。
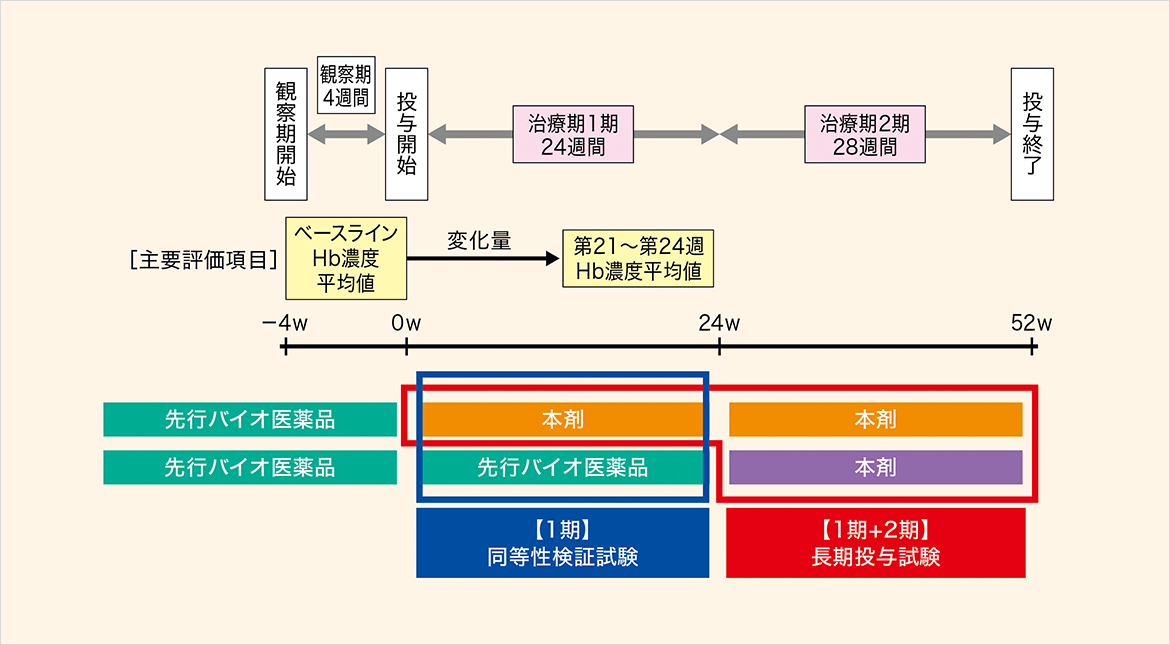
国内第Ⅲ相試験[血液透析(HD)患者対象、静脈内投与]
治療期1期(同等性検証試験)
有効性
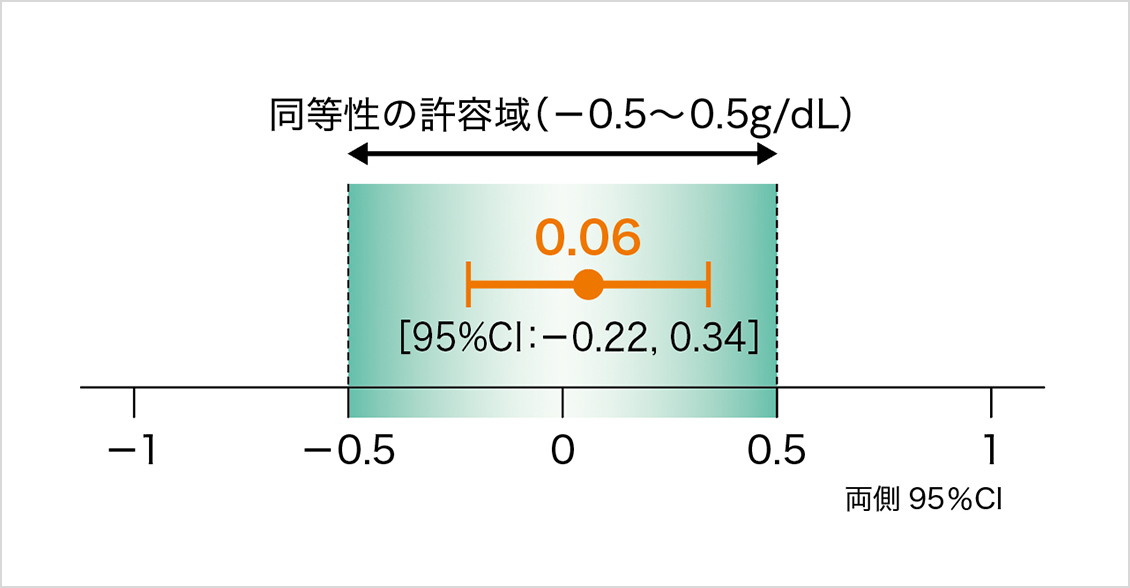
ヘモグロビン濃度変化量群間差の両側95%信頼区間(CI)が、同等性の許容域の範囲内であったことから、本剤と先行バイオ医薬品の同等性が検証された。
ヘモグロビン(Hb)濃度変化量[主要評価項目]
Hb濃度変化量(治療期1期 最終評価時)
(g/dl)
| 本剤群 (n=80) | 先行バイオ医薬品群 (n=87) | |
|---|---|---|
| ベースラインHb濃度※1 | 11.01±0.55 | 11.03±0.58 |
| 治療期第21~第24週のHb濃度※2 | 10.78±0.90 | 10.75±1.01 |
| Hb濃度の変化量※3 | -0.23±0.82 | -0.29±1.00 |
Hb濃度変化量の群間差(g/dL)
※1 観察期−3週から投与前まで4回の週初めの血液透析前Hb濃度の平均値
※2 治療期第21週から第24週まで4回の週初めの血液透析前Hb濃度の平均値
※3 治療期第21週から第24週まで4回のHb濃度の平均値※2とベースラインHb濃度※1の平均値の差
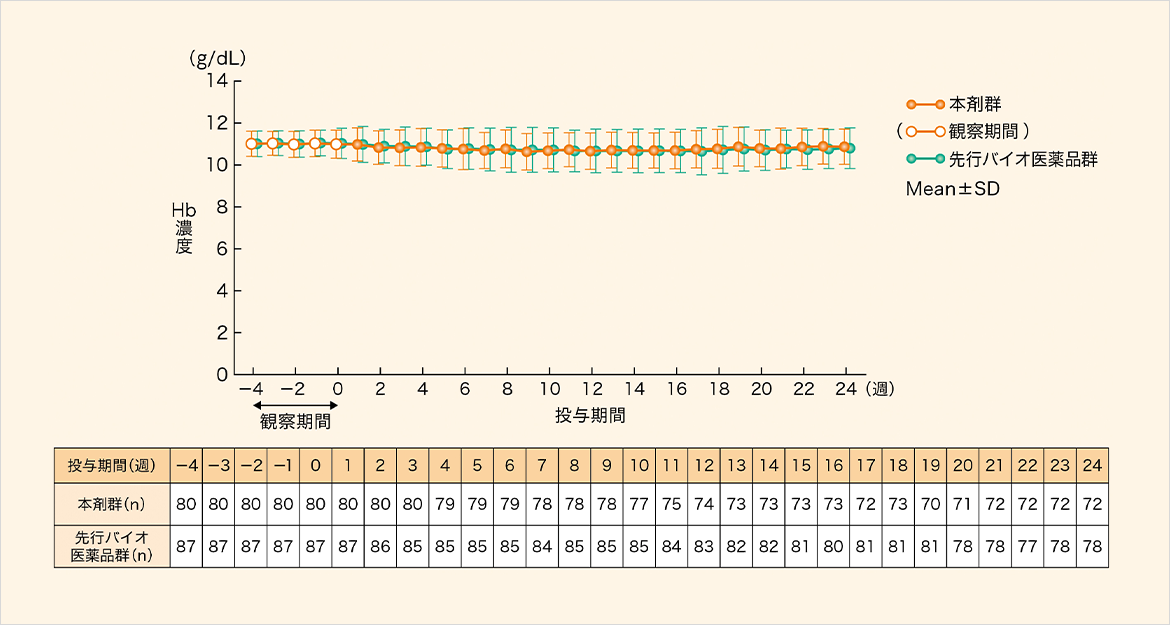
ヘモグロビン濃度の平均値は、本剤群では10.63~10.98g/dL、先行バイオ医薬品群では10.65~10.98g/dLの範囲で推移した。
ヘモグロビン(Hb)濃度推移[副次評価項目]
安全性
治療期1期における副作用は、本剤群では80例中2例(2.5%)、先行バイオ医薬品群では87例中2例(2.3%)に認められた。副作用としては、本剤群で急性心筋梗塞、脳梗塞及び高血圧が各1例(1.3%)に認められ、急性心筋梗塞と脳梗塞は同一症例であった。先行バイオ医薬品群で紅斑及び高血圧が各1例(1.1%)に認められた。死亡を含む重篤な副作用は本剤群で1例(2件)に認められ、事象は急性心筋梗塞及び脳梗塞であり、死亡例であった。副作用による中止例は本剤群で1例(2件)に認められ、重篤な副作用を発現した患者と同一症例であった。先行バイオ医薬品群では、死亡を含む重篤な副作用、及び副作用による中止例はいずれも認められなかった。
試験概要[治療期1期(同等性検証試験)]
- 目的
- 先行バイオ医薬品で治療中の腎性貧血を有するHD患者を対象に、本剤又は先行バイオ医薬品を24週間投与したときの同等性を、ヘモグロビン(Hb)濃度変化量を主要評価項目として検証した。また、安全性についても検討した。
- 対象
- 先行バイオ医薬品で治療中のHD患者167例[本剤群:80例、先行バイオ医薬品群:87例]
- 試験デザイン
- 多施設共同、実薬対照、ランダム化、単盲検(被験薬を扱う者と有効性及び安全性の評価に関わる者を別に設定し、被験者と有効性及び安全性の評価に関わる者に対して盲検とする)、並行群間比較
- 方法
- <用量及び投与方法>
観察期として先行バイオ医薬品を4週間静脈内投与し、その後、観察期の先行バイオ医薬品と同用量の本剤又は先行バイオ医薬品に切替え、週1回、週初めの血液透析日の血液透析終了時に透析回路の静脈側から24週間投与した。 - 評価項目
- 〈有効性評価項目〉
主要評価項目:治療期1期におけるHb濃度変化量(治療期第21週から第24週におけるHb濃度平均値と、ベースラインHb濃度平均値の差)
副次評価項目:Hb濃度推移(測定値及び変化量)、投与量の推移、総投与量、週あたりの平均投与量 等
※承認時評価資料
02 透析患者への有効性と安全性
国内第Ⅲ相試験[血液透析(HD)患者対象、静脈内投与]
治療期1期+2期(長期投与試験)
有効性
ヘモグロビン(Hb)濃度の平均値は、SS群とDS群の両群とも目標Hb濃度(10.0~12.0g/dL)の範囲内で推移した。
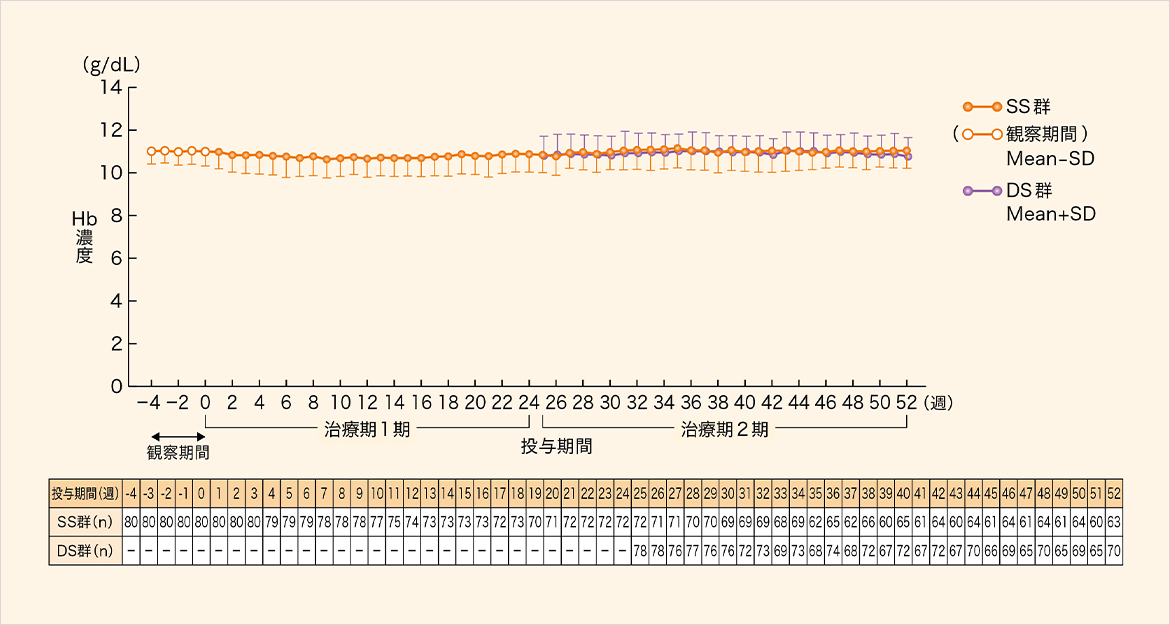
SS群:治療期1期...本剤投与、治療期2期...本剤投与
DS群:治療期1期...先行バイオ医薬品投与、治療期2期...本剤投与
安全性
治療期1期及び2期における本剤投与症例の副作用は、SS群では80例中3例(3.8%)、DS群では78例中2例(2.6%)に認められ、各副作用の発現は、下表の通りであった。
死亡を含む重篤な副作用はSS群で1例(2件)に認められ、事象は急性心筋梗塞及び脳梗塞で、死亡例であった(治療期1期に発現)。副作用による中止例はSS群で1例(2件)に認められ、重篤な副作用を発現した患者と同一症例であった。DS群では、死亡を含む重篤な副作用、及び副作用による中止例はいずれも認められなかった。
試験概要[治療期1期+2期(長期投与試験)]
治療期2期の試験方法で、治療期1期と異なっている部分について概要を以下に示す。
- 目的
- 治療期1期終了後、すべての症例に本剤を28週間投与したときの安全性及び有効性を検討した。
- 対象
- HD患者158例[SS群※1:80例、DS群※2:78例]
- 試験デザイン
- (治療期2期)多施設共同、非対照、非盲検
- 方法
- <用量及び投与方法(治療期2期)>
治療期1期の最終投与量と同用量の本剤を週1回、週初めの血液透析日の血液透析終了時に透析回路の静脈側から28週間投与した。 - 評価項目
- 有効性評価項目: Hb 濃度推移(測定値及び変化量)、投与量の推移 等
安全性評価項目:副作用 等
※1 SS群:治療期1期...本剤投与、治療期2期...本剤投与
※2 DS群:治療期1期...先行バイオ医薬品投与、治療期2期...本剤投与
※承認時評価資料
03 保存期慢性腎臓病(ND)患者への有効性・安全性
国内第Ⅲ相試験(長期投与試験)
[保存期慢性腎臓病(ND)の腎性貧血患者対象、皮下投与]
有効性
4週毎のヘモグロビン(Hb)濃度の平均値は目標Hb濃度(10.0~12.0g/dL)の範囲内で推移した。
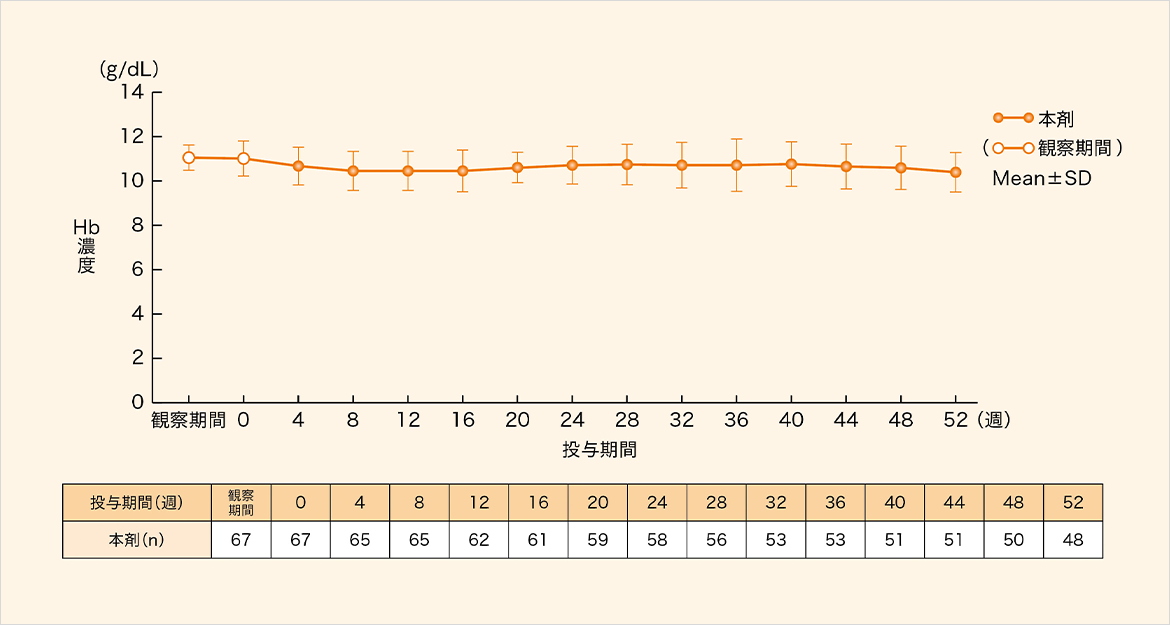
安全性
副作用は、67例中3例(4.5%)に認められた。副作用としては、網膜静脈閉塞、脳梗塞及び高血圧が各1例(1.5%)に認められた。死亡に至った副作用は認められなかった。その他の重篤な副作用は1例(1件)に認められ、事象は脳梗塞であった。副作用による中止例は1例(1件)に認められ、重篤な副作用を発現した患者と同一症例であった。
試験概要[治療期1期+2期(長期投与試験)]
- 目的
- 赤血球造血刺激因子製剤(ESA)で治療中のNDの腎性貧血患者を対象に、本剤に切替えて24週間及び52週間皮下投与した時の安全性及び有効性を検討した。
- 対象
- ESAで治療中のNDの腎性貧血患者67例
- 試験デザイン
- 多施設共同、オープン試験
- 方法
- <用量及び投与方法>
投与開始前のESAの用法及び用量を目安に本剤に切替え、2週に1回あるいは4週に1回、52週間、反復皮下投与した。投与開始時の投与頻度は、投与開始前に投与されたESAと同じ投与頻度とした。 - 評価項目
- 有効性評価項目:Hb濃度推移(測定値及び変化量)、投与量の推移 等
安全性評価項目:副作用 等
「禁忌を含む使用上の注意」等は電子添文をご参照ください
※承認時評価資料